
Large-scale / real materials simulations
Ab Initio Structure and Dynamics of CO2 at Supercritical Conditions
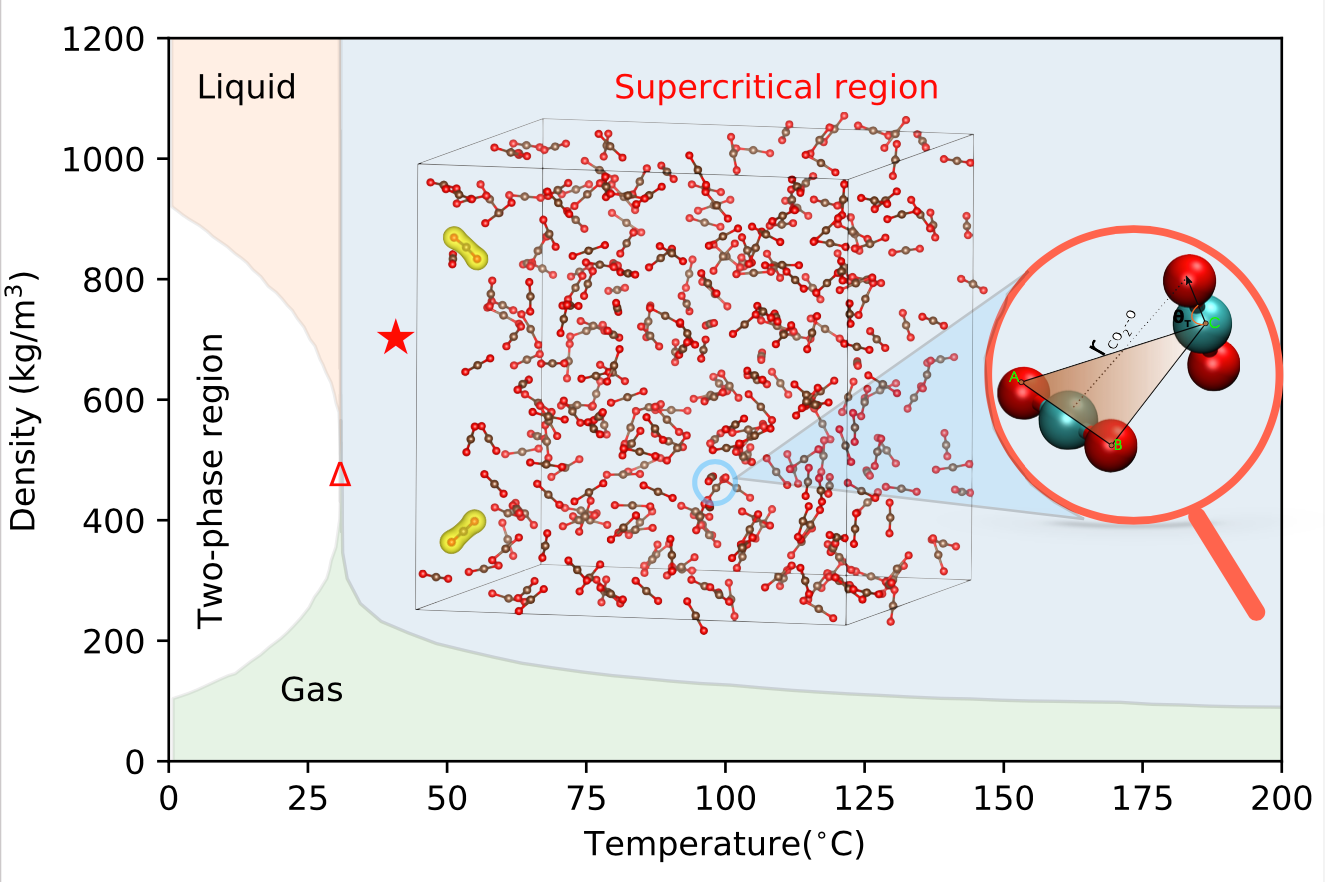
Green technologies rely on green solvents and fluids. Among them, supercritical CO2 already finds many important applications. The molecular-level understanding of the dynamics and structure of this supercritical fluid is a prerequisite for rational design of future green technologies. Unfortunately, the commonly employed Kohn–Sham density functional theory (DFT) is too computationally demanding to produce meaningfully converged dynamics within a reasonable time and with a reasonable computational effort. Thanks to subsystem DFT, we analyze finite-size effects by considering simulation cells of varying sizes (up to 256 independent molecules in the cell) and finite-time effects by running 100 ps trajectories. We find that the simulations are in reasonable and semiquantitative agreement with the available neutron diffraction experiments and that, as opposed to the gas phase, the CO2 molecules in the fluid are bent with an average OCO angle of 175.8°. Our simulations also confirm that the dimer T-shape is the most prevalent configuration. Our results further strengthen the experiment–simulation agreement for this fluid when comparing radial distribution functions and diffusion coefficien, confirming subsystem DFT as a viable tool for modeling structure and dynamics of condensed-phase systems.